1. グローバル医療機器市場の動向
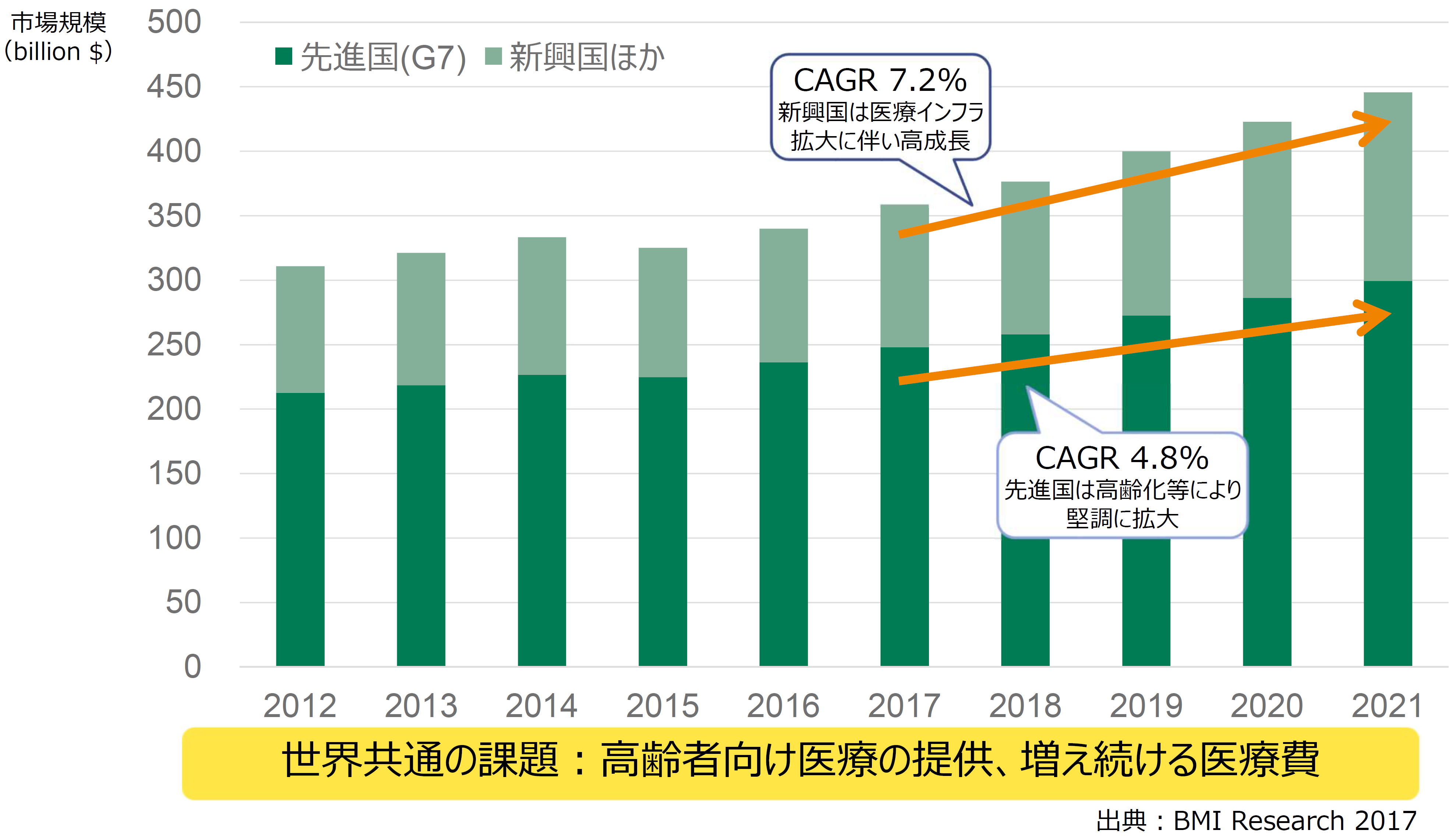
BMI Research(現Fitch Solutions, Inc.)による2017年の調査データ、並びに弊社調べにより、2017年の世界のヘルスケア市場は約165兆円[1]であり、その約1/4にあたる約40兆円[1]が医療機器市場と推測される。さらにその市場を地域別に見てみると、米国が43%、欧州が19%、日本と中国がそれぞれ7%、6%の市場規模を有している。市場成長率という点では、新興国では医療インフラの急速な拡大に伴い大きく拡大していくことが予想されており(CAGR=7.2%)、日米欧を中心とする先進国(G7)でも、高齢化などにより今後も市場は堅調に成長を継続するであろうと言われている(CAGR=4.8%)(図1)。弊社の売り上げの過去10年間の推移(図2)では、海外市場に注力してきた結果、国内での売り上げに比べ、海外での売り上げが急速に伸びていることがわかる。

2. 世界各国における薬事規制状況
新しい医療機器を患者様に届けるためには、多くの国で各国の規制に則った薬事承認/認証等(以下、薬事承認等)が必須であり、国連加盟国193か国のうち90か国以上において、なんらかの薬事規制が存在すると言われている[2]。薬事承認等とは、各医療機器のリスクの程度によって要求レベルは異なるが、安全性と有効性が各国の規制・基準に照らし合わせて審査され、各国の当局もしくは当局が指定した機関が承認/認証するプロセスを指す。薬事承認等に関する業務は開発した医療機器を患者様に届けるための最終段階のひとつであり、薬事承認等を取得することは開発者の立場から見ればゴールのひとつ、ビジネスの立場から見ればスタート地点と言えるであろう。
各国の薬事規制について考えるとき、医薬品については日米欧を中心とした各国で規制のハーモナイゼーションが進んでおり、医薬品規制調和国際会議(International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use, ICH)にて各種ガイドラインが作成され、薬事申請に必要な技術資料(Common Technical Document, CTD)の国際標準化等が進んでいる。医療機器においても、グローバルハーモナイゼーションタスクフォース(Global Harmonization task force, GHTF)や、その活動を引き継いだ国際医療機器規制当局フォーラム(International Medical Device Regulators Forum, IMDRF)のような世界各国の医療機器規制当局による任意の活動が、国際的な医療機器規制の整合化と収束を促進している。また、国を超えて薬事規制を共有する事例として、欧州連合加盟国を中心とした欧州34か国[3]にて本年5月26日より全面適用となる欧州医療機器規則(European Medical Device Regulation:EU-MDR)が挙げられる。そのほかにも、東南アジア諸国連合のASEAN医療機器指令(ASEAN Medical Device Directive, AMDD)や、ユーラシア経済連合(Eurasian economic union, EEU)においても規制のハーモナイゼーションに向けた取り組みが進められている。
一方、実際の医療機器における薬事承認等に関する業務を見てみると、米国、日本をはじめ、中国、韓国、インドなど多くの国ではそれぞれの国が有する独自の薬事規制に対応することが基本となっている。薬事承認等を得るための申請資料に含まれる情報としては、製品設計仕様、安全性、性能、有効性、製造に関する情報などが挙げられるが、どの情報が求められるのか、どれだけ詳細な情報が必要なのか、といったことは国によって要求が異なっている。さらに、参照すべき基準にその国独自の要求がある場合、追加のデータ収集が必要な場合もある。また、上述のASEANにおけるAMDDにおいても、関連国にて導入を進めてはいるものの、最終的にはそれぞれの国で独自の変更が加えられた上で、それぞれの国で薬事承認等を取得することが求められている。EEUにおける取り組みも、2021年12月31日までの移行期間が定められているが、その実現可能性はまだまだ不透明な状況である。規制のハーモナイゼーションが進められてはいるものの、実業務のレベルでは標準化が十分に進んでいるとは言い難い状況にある。
医療機器に関する規制は、その歴史が浅いことも特徴のひとつである。特にアジアや中南米、EMEA(Europe, Middle East and Africa)においてはこの10年で医療機器に関する薬事規制が新設された国が多い(マレーシア、パラグアイ、南アフリカなど)。さらに、中国やインドのように、既に薬事規制を有している国でも改訂が日々なされており、規制が強化されてきている。中国では2014年に医療機器規制の最上位に位置する医療機器監督管理条例が大きく改訂されたが、近い将来、更なる改訂が見込まれている。ここでは、薬事承認等取得にかかる要件の一部緩和が予定される一方、罰則規定などは強化される見込みである。また、インドではこれまで医療機器はDrugs and Cosmetics Rules, 1945を準用するかたちで規定されてきたが、2017年にDrugs and Cosmetics Rules, 1945に紐づくかたちでMedical Devices Rules, 2017が制定されており、新しい規制の下で再度薬事承認等の取得が求められている。国内においても、医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(医薬品医療機器法)の改正案が2019年11月に成立した。ガバナンスや安全対策等が強化されると共に、先駆け審査指定制度や条件付き早期承認制度など、革新的で医療上特に必要性が高い医療機器等への速やかな患者アクセスの確保を図る制度の法制化がなされた。
3. 医療機器のグローバル開発における薬事戦略
医療機器をグローバルに展開していくには、上述の通り、国毎に異なる薬事規制を理解すること、薬事規制の改訂や新規導入といった情報をタイムリーに入手すること、そしてこれらにスムーズに対応することが最重要であると考える。各国で求められる要求事項や薬事承認等にかかる期間等を把握し、効果的に薬事承認等を取得していくことが、グローバルでビジネスを拡大するための薬事戦略となり得るのである。実際、薬事承認等取得にかかる期間は、2ヵ月ほどの国から、準備期間も含めると2年以上を要する国もある。また、原産国での薬事承認等が必要な国や、日本・欧州・米国・カナダ・豪州いずれかで薬事承認等を取得していれば手続きが簡略化される国もある。例えば新しい医療機器を開発し、各国に順次上市していく場合、薬事承認等を取得する順番は各社の販売戦略によるところが大きいが、薬事承認等の観点を十分に取り込むことで、より効果的な戦略を立てることができると思われる。
各国の制度を利活用することも肝要である。例えば国内においては、上述の先駆け審査指定制度や条件付き早期承認制度は、医療上の必要性が高い医療機器や、希少疾病を治療する医療機器を早期に患者様のもとに届けるための制度であり、薬事承認等にかかる審査の短縮や必要なデータの軽減(ただし、市販後評価を拡充)が期待される。中国における国産医療機器優遇政策などもそのひとつと言えるであろう。
更には、広い視野に立ち、業界を通じて国に働きかけ、規制緩和や、他国とのハーモナイゼーション、2国間での薬事登録相互認証などを目的に、対話を進めてゆくことも薬事戦略のひとつとなり得る。これまでの行政における実績として、メキシコやインド、台湾の事例があるが、今後もより良い医療機器を患者様に早く届けるために、同様な取り組みに一企業として貢献していきたい。
4. 最後に
革新的な医療機器を開発したとしても、薬事承認等を取得できなければ、患者様に届けることはできない。一方で、各国の薬事規制は日々変化しており、また、厳格化されてきている。最新の薬事規制情報を入手、理解し、それらに効果的に対応することや、ローカルな国策を有効活用することが、グローバルにビジネスを維持・成長させるための戦略として重要である。
5. 参考
[1] 1$=110円換算
[2] 弊社調べ
[3] 欧州連合加盟国27か国、アイスランド、ノルウェー、リヒテンシュタイン、スイス、トルコ、マケドニアを含めた33か国に、今後の動向次第ではあるが、英国を加えて計34か国で予定されている。
(2020年2月22日受理, 2020年3月23日公開)

岩崎 一博 (いわさき かずひろ)
テルモ株式会社 レギュラトリーアフェアーズ 主任
略歴
2009年 九州大学理学部生物学科 卒業
2009年 テルモ株式会社入社 レギュラトリーアフェアーズ
2016年 テルモ株式会社 レギュラトリーアフェアーズ アジア担当主任
専門分野
医療機器薬事申請、グローバル薬事規制

千秋 和久 (せんしゅう かずひさ)
テルモ株式会社 執行役員 チーフクリニカル&レギュラトリーアフェアーズオフィサー
BMC33期
略歴
1987年 東京工業大学工学部高分子工学科卒業
1987年 テルモ株式会社入社 技術開発センター
1999年 工学博士取得(東京工業大学)
2009年 テルモヨーロッパ駐在 サイエンティフィッククリニカルマネージャー
2015年 テルモ株式会社 臨床薬事グローバル企画推進部長
2017年 テルモ株式会社 執行役員、チーフクリニカル&レギュラトリーアフェアーズオフィサー
専門分野
高分子化学、医療機器臨床薬事
